


Next: OBJECTIF DE LA THESE
Up: La génétique du psoriasis
Previous: PSORS5
Contents
La disponibilité de modčles animaux de maladies humaines, obtenus naturellement ou par ingénierie génétique en sur-exprimant dans des souris transgéniques, une protéine ou au contraire en détruisant le gčne avec déficience de la protéine correspondante permet de mieux comprendre les mécanismes biologiques impliqués et le rôle éventuel de la protéine impliquée dans la physiopathologie de la maladie. La reproduction des symptômes de la maladie dans ces modčles permet aussi de tester les traitements susceptibles de soigner la maladie chez l'homme.
Aucun modčle animal naturel n'existe pour le psoriasis. Il existe néanmoins des mutations spontanées ŕ l'état homozygote (Ttc7 , cpdm, Scd1
, cpdm, Scd1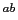 ) chez quelques souris qui entraînent des lésions dites psoriasiformes comme l'hyperprolifération épidermique, l'infiltration par des macrophages et des changements vasculaires. Cependant, ces souris ne montrent pas toutes les caractéristiques pathologiques du psoriasis comme la présence d'infiltration de lymphocytes T ainsi qu'une réponse aux traitements anti-psoriasis comme la cyclosporine et le calcipotriol [Gudjonsson et al., 2007].
) chez quelques souris qui entraînent des lésions dites psoriasiformes comme l'hyperprolifération épidermique, l'infiltration par des macrophages et des changements vasculaires. Cependant, ces souris ne montrent pas toutes les caractéristiques pathologiques du psoriasis comme la présence d'infiltration de lymphocytes T ainsi qu'une réponse aux traitements anti-psoriasis comme la cyclosporine et le calcipotriol [Gudjonsson et al., 2007].
Plusieurs modčles murins pour le psoriasis ont été développés, malgré des différences entre la peau et l'immunité humaine et murine (dont l'épaisseur et le nombre des couches de la peau). La plupart de ces modčles sur-exprime ou n'exprime plus des gčnes dont l'expression semble ętre altérée dans le psoriasis tels que les cytokines (IL-20, IL-6, IL-1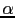 , IL-1ra) ou des facteurs de croissance (TGF-
, IL-1ra) ou des facteurs de croissance (TGF-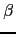 1, VEGF). Ces changements d'expression sont ciblés au niveau de la couche basale de l'épiderme grâce ŕ l'utilisation de promoteurs des gčnes de kératine KRT5 ou KRT14 ou au niveau de la couche suprabasale grâce aux promoteurs des gčnes KRT1, KRT10 ou de l'involucrine. L'ensemble des caractéristiques des modčles murins et leurs similarités avec le psoriasis humain sont détaillés dans le tableau 1.12Modčles murins et leur ressemblance au psoriasis humain.
1, VEGF). Ces changements d'expression sont ciblés au niveau de la couche basale de l'épiderme grâce ŕ l'utilisation de promoteurs des gčnes de kératine KRT5 ou KRT14 ou au niveau de la couche suprabasale grâce aux promoteurs des gčnes KRT1, KRT10 ou de l'involucrine. L'ensemble des caractéristiques des modčles murins et leurs similarités avec le psoriasis humain sont détaillés dans le tableau 1.12Modčles murins et leur ressemblance au psoriasis humain.
Les modčles murins semblent indiquer l'importance de la voie de Stat3, de NF-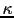 B, du facteur de transcription AP-1, des intégrines, des facteurs de croissance, des interleukines ainsi que des cellules T dans la survenue du psoriasis (Figure 1.15Trois voies de signalisation des modčles murins de psoriasis: STAT3, AP-1 et NF-$$B).
B, du facteur de transcription AP-1, des intégrines, des facteurs de croissance, des interleukines ainsi que des cellules T dans la survenue du psoriasis (Figure 1.15Trois voies de signalisation des modčles murins de psoriasis: STAT3, AP-1 et NF-$$B).
Stat3 est un facteur de transcription intervenant, quand il est activé, dans de nombreux processus physiologiques comme la prolifération, la migration et la survie cellulaire. Il semble aussi jouer un rôle dans le psoriasis. Des observations
Table 1.12:
Modčles murins et leur ressemblance au psoriasis humain
La premičre ligne indique les changements caractéristiques du psoriasis chez l'Homme suivi de ceux chez des modčles murins (Xg: xénogreffe, Alg: allogreffe avec des CMH différents, et Sp: modčles murins spontanés, Y, oui; N; non). Les différences avec l'Homme sont indiquées en rouge, les ressemblances en vert et les données inconnues en jaune. Les nombreux modčles murins ont la plupart des caractéristiques du psoriasis, malgré les différences histologiques de la peau des deux mammifčres. (D'aprčs [Gudjonsson et al., 2007])
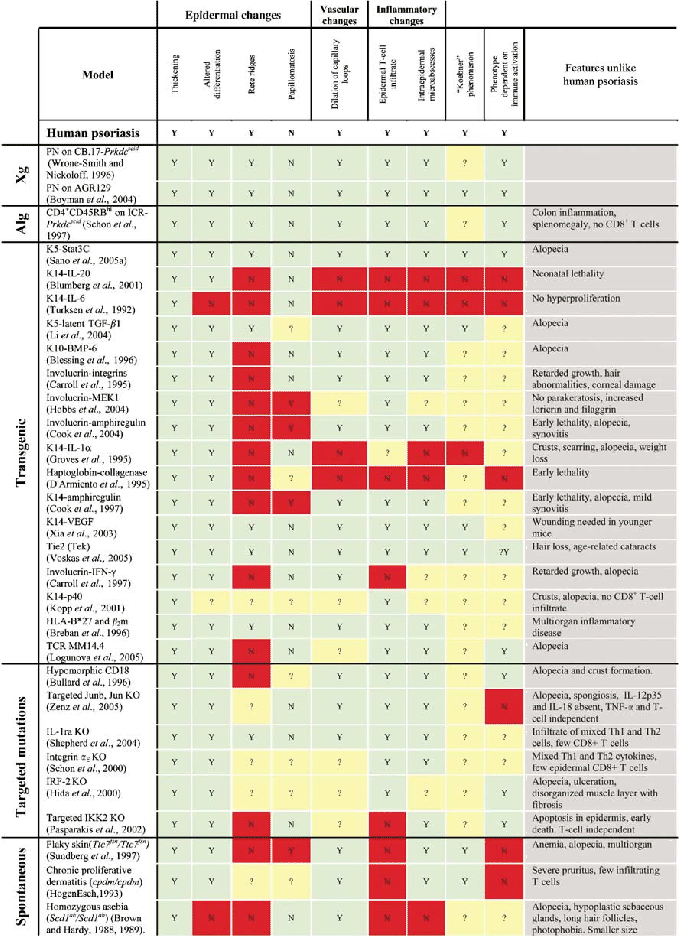 |
Figure 1.15:
Trois voies de signalisation des modčles murins de psoriasis: STAT3, AP-1 et NF-B
(a) Des signaux extracellulaires (IL-6, IL-20, etc) activent la voie STAT. Stat3 est activé par phosphorylation, suivi par sa dimérisation et sa translocation nucléaire. (b) Les protéines Jun (Jun, JunB, et JunD) avec d'autres protéines (Fos, ATF et CREB) sont les composants principaux de la famille du facteur de transcription AP-1. Jun intervient dans la prolifération cellulaire (via p53 et la cyclin D1), alors que JunB régule négativement la croissance cellulaire (via l'inhibiteur p16INK4a). (c) De nombreux signaux extracellulaires sont transmis au noyau via NF-B sous le contrôle du complexe kinase IkB (IKK). Activé lors de la phosphorylation de ses sous-unités IKKa et IKKb, IKK phosphorylise IkB, entraînant sa dissociation, suivi de sa dégradation. Ceci amčne ŕ la libération de NF-B actif (p50 and Rel-A(p65)) qui se transloque au noyau pour induire la transcription de gčnes cibles.
(D'aprčs [Gudjonsson et al., 2007])
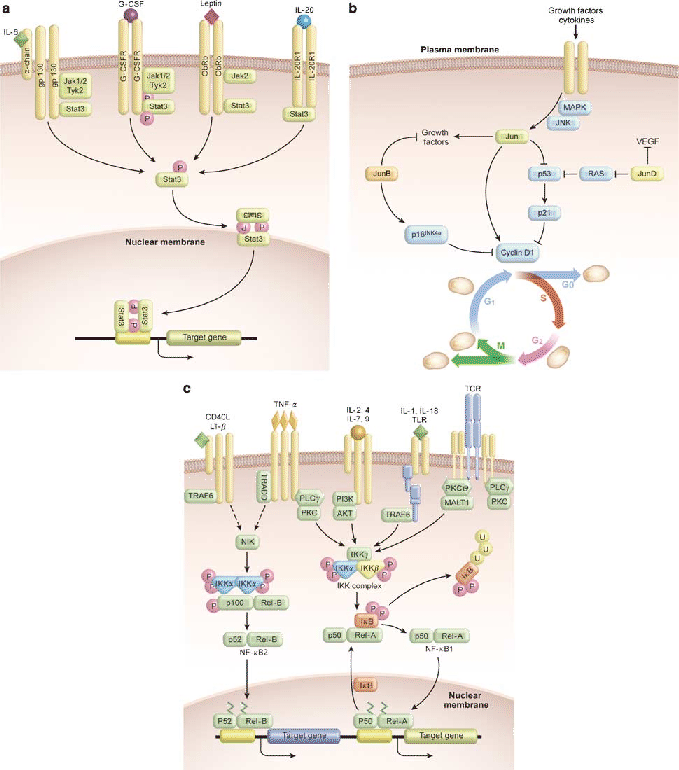 |
indiquent que Stat3 semble ętre un régulateur de la prolifération des kératinocytes et qu'il est présent sous forme activée au niveau des lésions psoriasiques humaines. Des lésions de la peau de type psoriasique ont d'ailleurs été obtenues spontanément ou en réponse ŕ une blessure chez une souris transgénique exprimant de maničre constitutive dans les kératinocytes basales Stat3 sous forme active. Cependant, le développement des lésions dans ce modčle semble ętre dépendant de l'activation des cellules T. D'autres souris transgéniques exprimant des activateurs potentiels de Stat3, montrés surexprimés dans la peau lésée, comme IL-20 et IL-6, montrent aussi des caractéristiques typiques du psoriasis telles que l'hyperprolifération des kératinocytes et une altération de la différenciation mais contrairement aux mutants Stat3, aucune infiltration par des leucocytes n'est observée.
Une autre voie semble intervenir dans la maladie: la voie NF-B. Cette voie de signalisation joue un rôle dans le contrôle de nombreux gčnes impliqués dans le développement, dans la mort cellulaire programmée mais surtout dans la réponse immune et inflammatoire et dans la prolifération cellulaire. Les facteurs de transcription NF-B sont maintenus ŕ l'état inactif par leur association avec les protéines inhibitrices de la famille des IB qui inhibent en partie la translocation au noyau de NF-B et la capacité de liaison de NF-B ŕ l'ADN. L'activation des dimčres NF-B est réalisée par le complexe IB kinase (IKK) qui est composé de deux sous-unités catalytiques IKK et IKK et d'une protéine régulatrice IKK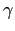 /NEMO. Lorsque IKK est activé, il phosphoryle IB qui conduit, lors de la fixation d'un complexe, ŕ la polyubiquitination de IB et son adressage vers le protéasome oů elle sera dégradée. Les dimčres NF-B sont alors transloqués dans le noyau oů ils activent la transcription de nombreux gčnes NF-B-dépendants. Des souris transgéniques avec délétion épidermique d'IKK ou d'IKK développent dans le premier cas une maladie de la peau inflammatoire et dans le second cas des anomalies de l'épiderme. Cependant, ces modčles montrent de grandes différences avec le psoriasis humain comme l'apoptose des kératinocytes pour les souris mutantes d'IKK [Gudjonsson et al., 2007].
/NEMO. Lorsque IKK est activé, il phosphoryle IB qui conduit, lors de la fixation d'un complexe, ŕ la polyubiquitination de IB et son adressage vers le protéasome oů elle sera dégradée. Les dimčres NF-B sont alors transloqués dans le noyau oů ils activent la transcription de nombreux gčnes NF-B-dépendants. Des souris transgéniques avec délétion épidermique d'IKK ou d'IKK développent dans le premier cas une maladie de la peau inflammatoire et dans le second cas des anomalies de l'épiderme. Cependant, ces modčles montrent de grandes différences avec le psoriasis humain comme l'apoptose des kératinocytes pour les souris mutantes d'IKK [Gudjonsson et al., 2007].
De maničre générale, les différents modčles murins ne possčde qu'une partie des caractéristiques de la maladie, ce qui indique une complexité de celle-ci et l'existence d'interaction entre différents types cellulaires et les cytokines dans la pathogenčse de la maladie.
En résumé, męme si les mécanismes physiopathologiques responsables du psoriasis restent encore inconnus, l'existence d'une composante génétique ŕ l'origine de la maladie faisant intervenir plusieurs gčnes et d'une composante environnementale est confirmée.
Next: OBJECTIF DE LA THESE
Up: La génétique du psoriasis
Previous: PSORS5
Contents
anouar
2009-08-22