Etudes de population ou cas-témoins
Un allèle est dit associé à une maladie s'il est plus fréquent parmi des malades non apparentés (ou les "cas") que parmi les témoins sains [Demenais et al., 1996,Feingold et al., 1998,Feingold, 2005,Monpetit and Chagnon, 2006]. Par opposition aux études de liaison génétique où la co-transmission d'une maladie et d'un marqueur au sein des généalogies est examinée, l'association maladie-marqueur est mise en évidence en comparant les fréquences alléliques du marqueur chez les sujets atteints et chez les témoins (non atteints). Un simple test de ![]() permet de tester la différence observée. Il est important que les deux groupes soient choisis au hasard d'une même population panmictique (les unions entre individus se font au hasard), sans critère de sélection autres que ceux correspondant à la maladie (l'âge et le sexe).
permet de tester la différence observée. Il est important que les deux groupes soient choisis au hasard d'une même population panmictique (les unions entre individus se font au hasard), sans critère de sélection autres que ceux correspondant à la maladie (l'âge et le sexe).
Une différence observée (association allélique positive) suggère que le marqueur est soit directement impliqué (et dans ce cas, cette association sera retrouvée dans différentes populations quelle que soit leur origine ethnique), soit en déséquilibre de liaison avec le(s) variant(s) causal(s).
Un déséquilibre de liaison (ou "DL") entre deux loci est défini par l'existence d'une combinaison d'allèles particulière à ces loci plus fréquente que celle attendue sous l'hypothèse d'association au hasard de ces allèles. En d'autres termes, dans le cas de deux allèles de deux loci, la fréquence des gamètes porteurs de ces deux allèles diffère de celle résultant du simple produit de leurs fréquences alléliques (voir ultérieurement Matériels et Méthodes).
En otant certains facteurs générateurs d'un DL entre loci tels que la migration ou le brassage des populations, deux marqueurs en fort déséquilibre de liaison (ou "déséquilibre gamétique") sont en général physiquement proches sur un même chromosome. Dans le cas de deux marqueurs en DL, la plupart des individus porteront la même combinaison de deux allèles spécifiques à ces marqueurs (un même haplotype). Généralement deux loci en déséquilibre de liaison seront également liés, mais l'inverse n'est pas nécessairement vrai. Un tel déséquilibre diminue au cours des générations successives entre autres par les événements de recombinaison et d'autant plus lentement que les deux loci sont très proches sur le même chromosome. Dans la plupart des cas, le marqueur dont un des allèles est trouvé associé à la maladie n'est pas souvent identifié comme le marqueur de prédisposition mais plutôt comme un marqueur en fort DL et proche du marqueur de susceptibilité à la maladie, surtout si peu de marqueurs ont été analysés lors de l'étude et s'il n'existe aucune preuve de son rôle dans la maladie sur le plan fonctionnel. L'existence d'un fort déséquilibre de liaison dépend en partie de l'histoire de la population étudiée. En effet, dans certains cas, le déséquilibre de liaison s'est constitué au sein d'une petite population "fondatrice" ou "isolée". La maladie serait due à une mutation apparue sur l'un des chromosomes d'un individu fondateur ou d'un nombre réduit de fondateurs (au maximum une centaine) et ainsi, les patients actuels partagent une toute petite région contenant cet allèle causal ancestral, variant selon la croissance de la population en question. Dans le cas d'une apparition relativement récente de l'allèle causatif d'une fréquence faible, le niveau du DL sera suffisamment élevé et étendu dans la région contenant le gène de prédisposition pour le détecter par ce type d'analyse. Si l'allèle à risque est fréquent dans la population actuelle (ce qui est attendu pour des maladies complexes), il a été sûrement associé à différents contextes chromosomiques dans la population ancestrale (conduisant à un plus faible DL dans la région concernée), rendant difficile sa détection par DL [Kruglyak, 1999].
D'une manière générale, le DL peut s'étendre sur différentes distances (de quelques kilobases à plusieurs centaines de kilobases), selon la population étudiée. Le génome est d'ailleurs organisé en blocs avec un fort DL. Ces blocs mesurent en moyenne 10 à 20 kb, mais varient en taille de quelques kilobases à plusieurs centaines de kilobases et sont séparés par des petites régions d'environ 1-2 kb comportant des taux de recombinaison élevés, dit "points chauds de recombinaison" car la recombinaison ne survient pas de façon aléatoire dans le génome [Monpetit and Chagnon, 2006]. Cinquante pour cent de la recombinaison totale survient dans 10 % de la séquence du génome. Par exemple, dans la région du complexe majeur d'histocompatibilité (CMH), c'est 80 % de la recombinaison qui est observée dans moins de 10 % de la séquence [McVean et al., 2004]. De plus, ces blocs de DL sont différents selon les populations. Par exemple, dans la
population humaine considérée comme la plus ancienne, la population africaine, le DL est moins fort et les blocs de DL observés s'étendent sur des distances plus courtes avec moins de SNPs, comparées aux populations européennes et asiatiques [Hinds et al., 2005]. Tous les humains descendent a priori d'ancêtres ayant vécu en Afrique il y a environ 150 000 ans. Ainsi, la plupart des variantes dans les populations actuelles, résultantes d'une migration, proviendraient d'une partie des variantes ayant existées dans cette population ancestrale. Contrairement aux autres populations, les populations d'Afrique ont été les plus nombreuses au cours du temps, multipliant d'autant leurs probabilités de scinder les haplotypes par recombinaison.
Grâce à différents projets d'analyse du génome humain (HapMap, Perlegen), la connaissance de l'ensemble de ces blocs dans différentes populations (caucasienne, africaine, asiatique) permet de réduire considérablement le nombre de SNPs à utiliser pour couvrir une région chromosomique ou le génome entier lors des études d'association. On estime que le nombre de SNPs nécessaires pour contenir la plupart de l'information sur la variation génétique du génome entier se situe entre 300 000 et 600 000. Pour ce dernier souvent utilisé dans le cas d'une étude sur des populations non-africaines, cela correspond à 1 SNP tous les 5kb.
Depuis les nombreux progrès technologiques, ce type d'analyse est maintenant réalisable et utilisé pour procéder à un criblage du génome entier, en analysant des centaines de milliers de SNPs répartis tout au long du génome. Cependant, il peut également être utilisé dans le cas où l'intervalle d'intérêt est restreint à quelques cM. En effet, puisque le nombre de gènes candidats dans cet intervalle est limité, une stratégie gène-candidat peut être réalisée en testant les gènes dont la fonction pourrait expliquer une partie de la maladie. Cette stratégie peut également être utilisée pour tester une région d'intérêt en analysant des marqueurs répartis uniformément le long de cette région, préférentiellement présents dans les
 |
gènes de l'intervalle. Comme discuté précédemment, le marqueur, qui est montré associé avec la maladie, peut être soit le marqueur causal soit être en déséquilibre de liaison avec le réel variant impliqué. Ainsi, dans la plupart des études sur des courtes régions (comme dans les "stratégies gènes candidats"), l'étude se focalise principalement sur les variants les plus susceptibles d'intervenir dans la prédisposition des maladies génétiques, comme par exemple, les variants présents dans la région codante qui peuvent avoir un rôle direct, plus ou moins connu, sur la fonction de la protéine correspondante. Cela a d'ailleurs été souvent le cas dans les maladies mendéliennes. Dans le cas des maladies complexes, les SNPs présents dans la région promotrice ou même dans la région non codante semblent avoir un rôle important dans le développement de ce type de maladie. Ainsi, afin de ne pas manquer le gène de prédisposition et le variant causal, il est important de prendre en compte l'existence du DL au sein du génome et de réaliser une approche plus générale. Souvent utilisée dans les études du criblage du génome, cette approche repose sur l'étude de variants pris sans a priori sur leur éventuel rôle fonctionnel, mais plutôt sélectionnés selon la structure du DL au sein de la région. Ainsi, même si le variant causal n'est pas testé, le gène sera quand même identifié car certains marqueurs de ce gène seront montrés associés avec la maladie, dû à l'existence d'un DL (complet ou non) entre ces marqueurs et le marqueur causal.
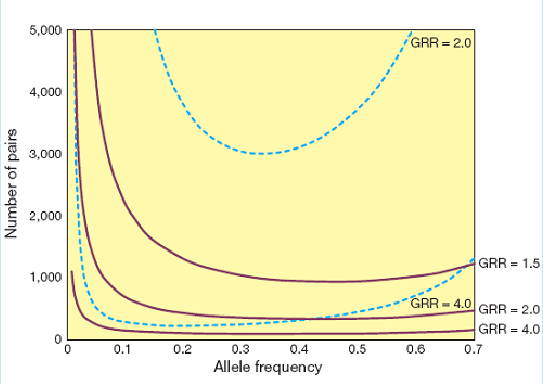
Cependant, la détection d'une association dépend, de manière générale, de l'intensité du DL entre le marqueur et la mutation causale, de la fréquence de la mutation dans la population, de l'effet de la mutation (sa pénétrance) sur la pathologie, de la distance génétique entre les marqueurs et le locus de la maladie, et enfin de l'hétérogénéité allélique. Ainsi, la taille de l'échantillon à tester doit être choisie, si possible, en conséquence.
Ce type d'analyse reste néanmoins plus puissant que les analyses de liaison pour l'identification des facteurs de prédisposition à des maladies complexes, même mineurs (Figure 1.7Comparaison entre les analyses de liaison et d'association pour détecter des effets génétiques) [Risch and Merikangas, 1996,Risch, 2000]. Il a d'ailleurs conduit à mettre en évidence le rôle du gène de l'apo-lipoprotéine E (APO E) comme facteur de risque dans la maladie d'Alzheimer et de celui de l'enzyme de conversion (ACE) dans l'infarctus du myocarde [Cambien et al., 1992,Saunders et al., 1993].
Avec ce type d'étude, il est également possible de calculer le risque relatif (odds-ratio, OR) de présenter la pathologie en fonction de l'état homozygote ou hétérozygote de l'allèle.
Néanmoins, cette méthode, facile à mettre en oeuvre, peut également induire en erreur. Une association observée peut ainsi résulter d'un artéfact dû à la structure de la population étudiée, une stratification de populations ou un mélange de populations ayant des fréquences différentes de la maladie et des allèles du marqueur (faux-positifs). En effet, il existe des disparités géographiques et ethniques des fréquences des marqueurs alléliques ou haplotypiques. Ainsi, il est important de sélectionner correctement la population témoin pour éviter de conclure faussement au rôle d'un marqueur et/ou d'un gène candidat.
Etudes familiales
Afin de contourner ce biais, des témoins "internes" pris au sein même des familles des malades ont été choisis. Les deux principales méthodes développées sur le principe de témoins "internes et fictifs" se font à partir de données familiales, les Trios (le patient et ses deux parents). Il s'agit du test d'haplotype à risque relatif (HRR, ou haplotype relative risk) et du test de transmission déséquilibrée (TDT, ou transmission disequilibrium test) [Demenais et al., 1996,Feingold et al., 1998,Feingold, 2005].
Le test HRR permet de comparer les fréquences alléliques du marqueur chez les patients et les témoins, où les témoins ne sont pas des individus ("témoins fictifs") mais reconstitués à partir des deux allèles parentaux non transmis aux patients. La signification est testée par un test de ![]() [Khoury, 1994].
[Khoury, 1994].
Le test du déséquilibre de transmission (TDT) compare les fréquences avec lesquelles un allèle particulier est transmis par un parent hétérozygote à l'enfant atteint. Il compare donc la fréquence de cet allèle parmi les allèles transmis et les allèles non transmis (définissant les "témoins fictifs"). La signification de l'association est testée par un test de ![]() [Spielman et al., 1993]. Si l'allèle est associé à la maladie, un excès de transmission de cet allèle sera observé dans plus de la moitié des cas. Ce test étudie à la fois l'association et la liaison génétique.
Cette méthode a été appliquée pour l'étude du gène de l'insuline dans le diabète insulino-dépendant [Julier et al., 1991].
[Spielman et al., 1993]. Si l'allèle est associé à la maladie, un excès de transmission de cet allèle sera observé dans plus de la moitié des cas. Ce test étudie à la fois l'association et la liaison génétique.
Cette méthode a été appliquée pour l'étude du gène de l'insuline dans le diabète insulino-dépendant [Julier et al., 1991].
La robustesse de ce test vis-à-vis des biais de stratification (migrations et/ou brassages des populations) se paie toutefois par une diminution de la puissance à détecter l'association puisqu'il est nécessaire que les parents soient hétérozygotes pour pouvoir réaliser ce test. De plus, ce test requiert le recrutement de familles Trio, ce qui est plus difficile à mettre en oeuvre que des individus non apparentés (comme pour une étude cas-témoins). Il nécessite en plus la connaissance des génotypes des parents, ce qui n'est pas accessible de manière systématique comme par exemple dans le cas de maladies à début tardif, telles que les maladies neurodégénératives. Des méthodes analogues ont été développées afin de se passer des données parentales (telles que le sib-TDT qui prend en compte les germains (atteints ou non) du patient) et/ou afin de considérer l'ensemble des apparentés pour reconstituer les données parentales (telles que le test basé sur les familles, FBAT (Family-based association test)).
Les analyses d'association sont réalisées avec différents programmes tels que GeneHunter [Kruglyak et al., 1996], Merlin [Abecasis et al., 2002], FBAT [Laird et al., 2000] selon le type de cohorte étudiée.
Quel que soit le test d'association utilisé, la question des faux-positifs engendrés par les tests multiples demeure : tester n marqueurs avec k allèles revient à réaliser environ n(k-1) tests indépendants. La signification de chaque test doit, en toute rigueur, être divisée par ce facteur (correction de Bonferroni). Par exemple, une valeur de signification nominale de p=0.0005 est requise pour atteindre un simple taux de 5% de faux positifs (l'erreur de type I) lorsque l'on teste indépendamment 100 polymorphismes bialléliques sur des gènes différents. Cette correction est cependant très conservatrice, surtout dans le cas des maladies multifactorielles où les nombreux tests réalisées lors des études ne sont pas souvent indépendants. Par exemple, les marqueurs étudiés sont souvent dépendants entre eux par l'existence d'un DL entre ces marqueurs. De plus, parmi les nombreux facteurs connus pour intervenir dans la puissance de l'étude, le facteur "taille de l'échantillon" doit être suffisamment important pour détecter l'ensemble des loci (faux-négatifs), ce qui est souvent peu réalisable en pratique. Ainsi, il est préférable d'être moins stringent vis à vis du seuil limite de détection, en tolérant un seuil de faux-positifs élevé. Une autre approche consiste à utiliser le test de permutations avec des séries de données générées par une simulation, pour établir la répartition attendue des faux-positifs et ajuster les seuils en conséquence. La réplication des résultats d'une étude par d'autres études indépendantes, en complément des données de liaison, reste le plus souhaitable pour garantir l'implication d'un locus ou d'un gène candidat car en général, la mise en évidence d'une association ne permet pas de conclure à l'implication formelle du gène candidat testé dans la susceptibilité génétique de la maladie, mais seulement de l'existence d'un facteur génétique à proximité (quelques kilobases). En effet, si le marqueur associé est directement impliqué dans la maladie, il sera trouvé associé dans différentes populations quelle que soit leur origine ethnique et leur structure de DL, variable selon leur histoire. Cependant, ce sont les études fonctionnelles sur les variants associés, si cela est possible, qui peuvent démontrer le mécanisme biologique en cause dans la maladie.