Le but des analyses de liaison (paramétriques ou non paramétriques) est de localiser les régions contenant les gčnes responsables du trait ou de la maladie sur le génome au moyen d'observations sur des individus reliés. La méthode des lod-scores est basée sur la co-ségrégation, au cours des générations, des allčles de marqueurs polymorphes avec le locus responsable de la maladie (ou du trait), dans une ou un échantillon de familles [Demenais et al., 1996,Feingold et al., 1998,Campion, 2001]. Cette co-ségrégation est due ŕ l'existence d'une liaison génétique entre ces deux loci (gčne-marqueur). Deux loci (marqueur-marqueur/ gčne-marqueur) sont dits "liés" s'ils sont suffisamment proches l'un de l'autre sur le męme chromosome pour ętre cohérités dans plus de 50% des cas. Cette liaison est dépendante d'un événement biologique de la méiose qui casse la co-ségrégation, la recombinaison intra-chromosomique ("crossing-over"). Deux loci sont rarement disjoints par un événement de recombinaison si la distance physique entre les deux loci est suffisamment faible. L'analyse de liaison repose donc sur la fraction de recombinaison ![]() (la probabilité de recombinaison entre deux loci ŕ la méiose) qui est fonction de la distance entre les loci. Cette distance est exprimée en unité de recombinaison ou centiMorgan : 1 cM correspond ŕ une fréquence de recombinaison de 1% entre les deux loci. Si
(la probabilité de recombinaison entre deux loci ŕ la méiose) qui est fonction de la distance entre les loci. Cette distance est exprimée en unité de recombinaison ou centiMorgan : 1 cM correspond ŕ une fréquence de recombinaison de 1% entre les deux loci. Si ![]() =0, les deux loci sont co-hérités dans 100% des cas (liaison parfaite). La distance génétique entre les deux loci est alors de 0 cM. Si
=0, les deux loci sont co-hérités dans 100% des cas (liaison parfaite). La distance génétique entre les deux loci est alors de 0 cM. Si ![]() =0.5, les deux loci sont génétiquement indépendants en termes de ségrégation. Les deux loci se situent soit sur deux chromosomes différents soit sont ŕ une distance suffisamment grande sur le męme chromosome (>50 cM). L'analyse de liaison repose sur le logarithme du rapport de vraisemblance entre deux hypothčses (l'hypothčse ŕ tester versus l'hypothčse nulle). Le degré de la liaison génétique, exprimée en lod score (Z) par Morton (1955), est obtenue par le logarithme décimal du rapport entre la vraisemblance de liaison (l'hypothčse alternative) entre le marqueur (position connue) et le gčne de la maladie (position inconnue) pour une distance génétique donnée, représentée par le taux de recombinaison
=0.5, les deux loci sont génétiquement indépendants en termes de ségrégation. Les deux loci se situent soit sur deux chromosomes différents soit sont ŕ une distance suffisamment grande sur le męme chromosome (>50 cM). L'analyse de liaison repose sur le logarithme du rapport de vraisemblance entre deux hypothčses (l'hypothčse ŕ tester versus l'hypothčse nulle). Le degré de la liaison génétique, exprimée en lod score (Z) par Morton (1955), est obtenue par le logarithme décimal du rapport entre la vraisemblance de liaison (l'hypothčse alternative) entre le marqueur (position connue) et le gčne de la maladie (position inconnue) pour une distance génétique donnée, représentée par le taux de recombinaison ![]() (0
(0
![]() 0.5) et l'hypothčse d'indépendance génétique entre ce marqueur et le gčne recherché (hypothčse nulle,
0.5) et l'hypothčse d'indépendance génétique entre ce marqueur et le gčne recherché (hypothčse nulle, ![]() =0.5): Z(
=0.5): Z(![]() )=
)= ![]() [L(
[L(![]() )/L(0.5)] [Morton, 1955]. L'analyse de liaison teste la vraisemblance d'une liaison génétique pour diverses valeurs de recombinaison
)/L(0.5)] [Morton, 1955]. L'analyse de liaison teste la vraisemblance d'une liaison génétique pour diverses valeurs de recombinaison ![]() voisines de zéro et pour un marqueur donné. Le lod score varie donc suivant la valeur de
voisines de zéro et pour un marqueur donné. Le lod score varie donc suivant la valeur de ![]() considérée. Plus le lod score est élevé, plus la probabilité de la liaison entre le marqueur et le gčne causal est grande et inversement. Classiquement, l'hypothčse d'indépendance est rejetée avec un score Z(
considérée. Plus le lod score est élevé, plus la probabilité de la liaison entre le marqueur et le gčne causal est grande et inversement. Classiquement, l'hypothčse d'indépendance est rejetée avec un score Z(![]() ) supérieur ŕ 3 (correspondant ŕ P=10
) supérieur ŕ 3 (correspondant ŕ P=10![]() ). Il y a alors liaison génétique (ou "linkage" en anglais) entre le marqueur et la maladie. L'hypothčse de liaison est rejetée si Z(
). Il y a alors liaison génétique (ou "linkage" en anglais) entre le marqueur et la maladie. L'hypothčse de liaison est rejetée si Z(![]() )<-2. Cela signifie qu'il y a une preuve que des événements de recombinaison ont eu lieu entre le marqueur et le locus maladie. Pour un lod score entre ces deux valeurs, on ne peut pas conclure ni ŕ une liaison ni ŕ une exclusion, il faut analyser d'autres familles. Dans le cas de stratégie "genome-wide scan", le seuil monte ŕ 3.3 pour conclure ŕ une liaison avec une valeur de P de 0.05 [Lander and Kruglyak, 1995].
)<-2. Cela signifie qu'il y a une preuve que des événements de recombinaison ont eu lieu entre le marqueur et le locus maladie. Pour un lod score entre ces deux valeurs, on ne peut pas conclure ni ŕ une liaison ni ŕ une exclusion, il faut analyser d'autres familles. Dans le cas de stratégie "genome-wide scan", le seuil monte ŕ 3.3 pour conclure ŕ une liaison avec une valeur de P de 0.05 [Lander and Kruglyak, 1995].
L'objectif final de l'analyse est d'estimer la fraction de recombinaison (ou la position chromosomique (cM)) entre chaque marqueur et le locus de la maladie (analyse bi-point) ou la position du locus de la maladie relative ŕ une carte fixe des marqueurs oů l'on suppose que l'endroit de chaque marqueur est connu (analyse multipoint). L'analyse multipoint peut ętre plus puissante car l'informativité d'un marqueur peut ętre améliorée en le considérant simultanément avec les marqueurs voisins. Elle permet aussi d'avoir une estimation de la localisation du locus "maladie" et de se protéger contre les résultats abérrants obtenus pour un marqueur. Cependant, elle reste sensible ŕ la présence d'erreurs de génotypage et elle requiert un ordre correct des marqueurs ainsi que de la distance entre les marqueurs. La meilleure estimation (le maximum de vraisemblance) de ![]() ou de la position est celle qui maximise la fonction du lod score: le lod score maximum. De plus, męme si cette analyse est basée sur l'existence d'un locus majeur ŕ l'origine du trait, elle peut également prendre en compte l'hétérogénéité génétique ŕ l'aide du paramčtre
ou de la position est celle qui maximise la fonction du lod score: le lod score maximum. De plus, męme si cette analyse est basée sur l'existence d'un locus majeur ŕ l'origine du trait, elle peut également prendre en compte l'hétérogénéité génétique ŕ l'aide du paramčtre ![]() qui répartit les familles en familles liées et non liées au marqueur considéré.Le lod score est alors maximisé en fonction des paramčtres
qui répartit les familles en familles liées et non liées au marqueur considéré.Le lod score est alors maximisé en fonction des paramčtres ![]() et
et ![]() (la proportion de familles liées au locus étudié). Ce lod score, intitulé "HLOD" (heterogeneity LOD) est défini comme étant:
(la proportion de familles liées au locus étudié). Ce lod score, intitulé "HLOD" (heterogeneity LOD) est défini comme étant:
![]() =
=![]() [L(
[L(![]() )/L(
)/L(![]() =1,
=1,![]() =0.5)].
=0.5)].
Les analyses de liaison paramétrées sont réalisées avec différents programmes comme Linkage [Lathrop et al., 1984], GeneHunter [Kruglyak et al., 1996], Merlin [Abecasis et al., 2002], Simwalk2 [Sobel and Lange, 1996], qui utilisent divers algorithmes (Elston-Stewart, Lander-Green, chaines de Markov Monte Carlo) pour permettre d'étudier de nombreux marqueurs dans des familles de toutes tailles.
Cette méthode est trčs puissante surtout lors de son application ŕ l'étude de plusieurs grandes généalogies oů un męme gčne majeur, quels que soient les allčles impliqués, est ŕ l'origine de la maladie. En effet, dans les études de liaison génétique, l'existence d'une hétérogénéité allélique n'affecte pas l'analyse car toutes les familles (avec différentes mutations) montreront une liaison avec la męme région chromosomique. Selon les familles, différents allčles d'un męme marqueur seront alors liés ŕ la maladie. Elle fut d'ailleurs largement utilisée pour cartographier de nombreux gčnes impliqués dans des maladies mendéliennes. Dans ce cas, l'étude de grandes familles est vraiment intéressante car il y a une trčs forte probabilité pour que l'ensemble des cas développés au sein d'une męme famille soit dű ŕ une susceptibilité commune. Cependant, cette méthode reste peu adaptée dans le cas des maladies complexes avec hétérogénéité non allélique. Etant une méthode paramétrée, elle nécessite la connaissance du modčle génétique (mode de transmission) de la maladie, c'est-ŕ-dire si la maladie est autosomique ou liée au chromosome X, si elle est dominante ou récessive. Elle dépend de la fréquence et de la pénétrance de l'allčle de prédisposition ŕ la maladie, de la fréquence des phénocopies, du taux de mutation et de la fréquence allélique des marqueurs de la maladie. Ces paramčtres sont souvent préalablement estimés par les études de ségrégation. De maničre générale, elle dépend aussi de la famille étudiée, sa taille et sa composition en nombre d'individus atteints, et des marqueurs microsatellites selon leur informativité (degré de polymorphisme). Dans le cas des maladies complexes, le mode héréditaire de transmission de la maladie est souvent trop imprécis. La méconnaissance de ces paramčtres conduit généralement ŕ une perte de puissance pour détecter une liaison existante (faux négatifs) et/ou ŕ détecter par hasard une liaison inexistante (faux positifs). A moins d'avoir montré lors d'une analyse de ségrégation l'implication d'un gčne majeur, il serait peu adapté d'utiliser cette méthode dans l'étude des maladies complexes.
Analyse de liaison non paramétrique ou méthode des paires de
germains
Pour les raisons précédemment décrites, d'autres méthodes, dites non paramétri-
 |
ques (ou "model-free") peuvent ętre utilisées [Demenais et al., 1996,Seboun, 1996,Feingold et al., 1998,Campion, 2001,Feingold, 2005]. Elles ne font aucune hypothčse a priori sur le mode héréditaire de transmission de la maladie. Une des méthodes est également une analyse de liaison qui repose sur le principe du partage d'allčles. Elle tente ŕ montrer que pour des apparentés qui se ressemblent pour un trait (la maladie), la transmission d'un ou des segments chromosomiques ne correspond pas ŕ celle obtenue lors d'une ségrégation aléatoire mendélienne mais qu'au contraire ces apparentés héritent d'un męme segment chromosomique parental plus souvent que ne le voudrait le hasard. En d'autres termes, si un marqueur analysé est proche du locus du gčne incriminé, les apparentés atteints auront tendance ŕ partager le męme segment et donc les męmes allčles ŕ ce site. Ces apparentés présenteront donc un excčs d'allčles du marqueur en commun.
Selon ce principe, la méthode a été appliquée sur les paires de germains atteints de la męme maladie (Affected Sibling Pairs, ASP)[Penrose, 1935].
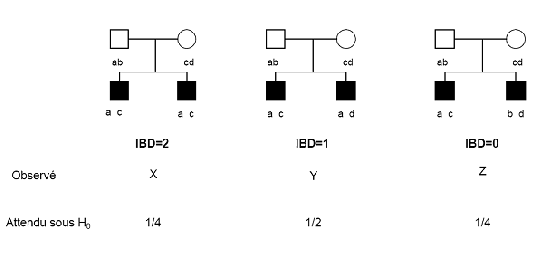
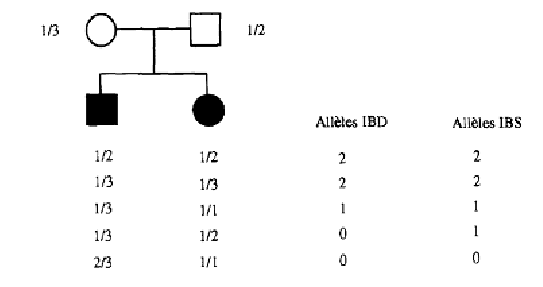
Cette méthode des paires de germains se base sur le fait que s'il existe une liaison, une transmission non aléatoire des allčles parentaux est observée chez les enfants atteints. Dans ce cas, les enfants atteints héritent plus souvent des męmes allčles du locus considéré que ne le voudrait le hasard. L'allčle commun entre deux germains provenant d'un męme allčle parental est dit "identique par descendance" (idendical by descent, IBD) [Campion, 2001,Feingold, 2005]. Cette méthode évalue, pour un marqueur donné, la proportion du nombre d'allčles identiques par descendance au sein de paires de patients. Ainsi, pour un marqueur, les germains peuvent avoir en commun 2, 1 ou 0 allčles IBD hérités de leurs parents. Les germains qui partagent deux allčles IBD (IBD2) ont hérité des deux męmes allčles de leurs deux parents. S'ils partagent un allčle IBD (IBD1), ils ont hérité du męme allčle d'un męme parent. Et enfin, ils ne partagent aucun allčle IBD (IBD0) s'ils héritent d'allčles différents. Dans le cas d'une ségrégation mendélienne (en absence de liaison), les germains vont hériter de 2 allčles IBD d'un locus donné dans 25% des cas, d'un allčle IBD dans 50% des cas, et de 0 allčles IBD dans les 25% restants. Ces distributions attendues sont alors comparées aux distributions observées par un simple test de ![]() qui devient significatif en cas de liaison (Figure 1.5Différents états IBD possibles pour une paire de germains atteints). Un autre test, plus puissant qui teste spécialement l'augmentation du partage est le 'test moyen' [Blackwelder and Elston, 1985]. Pour tous ces tests, l'augmentation du partage des allčles identiques par descendance chez les paires de germains indique une liaison génétique entre le marqueur et le gčne de prédisposition.
qui devient significatif en cas de liaison (Figure 1.5Différents états IBD possibles pour une paire de germains atteints). Un autre test, plus puissant qui teste spécialement l'augmentation du partage est le 'test moyen' [Blackwelder and Elston, 1985]. Pour tous ces tests, l'augmentation du partage des allčles identiques par descendance chez les paires de germains indique une liaison génétique entre le marqueur et le gčne de prédisposition.
Ne requérant aucune hypothčse sur le modčle génétique de transmission, cette approche est largement utilisée dans le cas d'études de maladies complexes. Cette approche a permis d'ailleurs de mettre en évidence la liaison du systčme HLA avec différentes maladies auto-immunes, telles que la sclérose en plaques ou le diabčte insulinodépendant (DID) [Demenais et al., 1996]. Męme si cette méthode
 |
Les méthodes non paramétriques ont également été utilisées pour des analyses de liaison ŕ un trait quantitatif (QTL, Quantitative Trait Loci). Proposée par Haseman et Elston (1972), cette approche est basée sur la corrélation entre le statut IBD des germains ŕ un marqueur et leur ressemblance phénotypique (trait clinique ou biologique), mesurée par la différence au carré entre les valeurs quantitatives du trait [JK. and RC., 1972]. Plusieurs variantes de cette méthode existent dont une fondée sur la décomposition de la variance.
Les analyses de liaison non paramétrées sont réalisées entre autres avec différents programmes tels que GeneHunter [Kruglyak et al., 1996], Merlin [Abecasis et al., 2002] ou SIMWALK2 [Sobel and Lange, 1996].
Enfin, la méthode générale des paires de germains n'est généralement pas capable de localiser un gčne avec précision. En effet, le manque de puissance de cette méthode pour la détection de gčnes ayant un effet mineur entraîne souvent la détection de fausses régions de prédisposition (faux-positifs), pouvant expliquer le peu de réplication des loci initialement suggérés. Et męme si ces méthodes non paramétriques ont permis d'identifier surtout des régions d'intéręt contenant des gčnes ŕ effet fort, les régions identifiées ne sont souvent pas suffisamment petites (10 ŕ 30 cM) pour permettre le clonage positionnel facile du gčne causal, quelles que soient les approches utilisées (criblage systématique du génome ou cartographie fine) [Risch and Merikangas, 1996]. L'approche suivante sera alors la recherche d'association allélique (ou d'un ensemble d'allčles ("association haplotypique")), dans la population générale, entre la maladie et les allčles de marqueurs bi-alléliques présents soit dans les régions de prédisposition pour une cartographie fine, ou soit dans les gčnes candidats pour une stratégie gčne-candidat. Elle peut également ętre utilisée comme alternative des analyses de liaison lors d'un criblage du génome, avec des SNPs répartis sur le génome entier.