Les membres principaux du complexe AP-1 sont les protéines de la famille de Jun (Jun, JunB et JunD), de Fos, de ATF et de fibrosarcomes musculoaponeurotiques (Maf). Les protéines Jun et Fos fonctionnent comme des facteurs de transcription dimériques. Sous forme d'homodimères et /ou d'hétérodimères, ils se lient à l'ADN au niveau des sites de liaison d'AP-1 présents dans les régions "enhancer" et "promotrices" de nombreux gènes mammifères cibles pour favoriser leur transcription [Zenz and Wagner, 2006]. Les différents membres des familles de Jun et de Fos montrent des différences significatives dans leur activité transcriptionnelle et dans la liaison à l'ADN suggérant ainsi des fonctions spécifiques dans la régulation des gènes cibles pour les différents dimères d'AP-1 [Shaulian and Karin, 2002]. Dans le cas de Jun et de JunB, ils diffèrent considérablement dans leur capacité à réguler de gènes cibles. Par suite de la faible affinité de JunB pour s'homodimériser ainsi que pour se lier à l'AP-1, JunB a besoin d'interactions synergiques avec de nombreux homodimères pour activer efficacement les promoteurs. Contrairement à JunB, Jun nécessite un seul site de liaison au complexe AP-1 pour intervenir dans la transcription des gènes [Shaulian and Karin, 2002,Zenz and Wagner, 2006]. De plus, ces protéines montrent des fonctions antagonistes. Par exemple, dans le cas de la prolifération et de la différenciation des kératinocytes lors du développement et lors du cancer de la peau, Jun est considéré comme un régulateur positif de la prolifération par son effet transcriptionnel direct sur l'expression épidermique du récepteur de facteur de croissance (EGFR). En revanche, JunB peut empêcher la prolifération des kératinocytes et des cellules souches hématopoïétiques. En plus de son implication dans la balance des processus de prolifération et de différenciation des kératinocytes, le complexe AP-1 intervient aussi sur de nombreux gènes clés de l'homéostasie de la peau, tels que les gènes codants pour les tranglutaminases, les cytokératines, la profilagrine, et sur des gènes impliqués dans l'inflammation comme les cytokines et leurs récepteurs (TNF![]() , IL-8, IL-1RN), les facteurs de croissance [Zenz and Wagner, 2006].
, IL-8, IL-1RN), les facteurs de croissance [Zenz and Wagner, 2006].
Chez l'homme, quelques études ont suggéré un rôle de ces protéines dans l'étiologie du psoriasis. En effet, une étude d'expression de JunB et de Jun, dans des biopsies de peau de zones non lésées et lésées de 8 patients atteints de psoriasis, montre que dans l'épiderme non atteint des patients, JunB est exprimé dans toutes les couches de l'épiderme, avec des niveaux plus élevés dans les couches basales et spineuses [Zenz et al., 2005]. Ce profil est semblable à celui observé lors de l'étude de la peau saine. En revanche, l'expression de JunB est considérablement réduite dans la peau lésée de patients atteints d'une forme grave de psoriasis (n=6) et intermédiaire dans celle de patients atteints d'une forme modérée de psoriasis (n=2), suggérant un rôle possible de JunB dans le développement de la maladie. Jun, un antagoniste de JunB, est seulement faiblement exprimé dans l'épiderme normal, excepté dans la couche granulaire. Contrairement à JunB, l'expression de Jun est augmentée dans la peau lésée. Ainsi, une réduction forte du rapport JunB/ Jun est observée dans la peau lésée, surtout dans la couche basale, ce qui pourrait expliquer le défaut de différenciation des kératinocytes [Zenz et al., 2005]. Une étude fonctionnelle, sur un modèle murin de Knock-out inductible des protéines Jun/JunB dans les kératinocytes semble confirmer le rôle de ces protéines dans le psoriasis. Dans le cas des composants d'AP-1, la létalité embryonnaire des divers knock-outs des éléments d'AP-1, par exemple pour Jun, JunB, Fra-1 et Fra-2, empêche en grande partie des études fonctionnelles in vivo mais montrent l'importance de ces gènes dans le développement embryonnaire. Ainsi, les stratégies de knock-outs conditionnels et inductibles chez la souris, en particulier dans l'épiderme, sont devenues un modèle important pour étudier la régulation et la fonction des sous-unités AP-1 dans des processus physiologiques et pathologiques in vivo. Dans un délai de 2 semaines, contrairement aux délétions spécifiques à l'épiderme de seulement JunB ou Jun chez les souris transgéniques, un phénotype, ressemblant fortement aux lésions des patients atteints de psoriasis, tels que des plaques écailleuses affectant principalement les oreilles, les pattes et la queue, et moins fréquemment la peau arrière velue et des altérations de la peau "chauve", est observé lors de la double-délétion de JunB et de Jun propre à l'épiderme et inductible chez les souris transgéniques. D'autres caractéristiques du psoriasis sont présentes, telles que l'épaississement de l'épiderme, l'épaississement des couches supérieures kératinisées avec la parakératose (les kératinocytes nucléés dans la couche cornifiée) et l'augmentation de la vascularisation sous-épidermique. Des cellules T intra-épidermiques et des cellules inflammatoires typiques (neutrophiles et macrophages) sont présentes dans le derme et au niveau des articulations. Enfin, un grand nombre de cytokines, de chémokines et de facteurs de transcription, éléments contribuant sûrement à la pathogénie du psoriasis, s'avèrent être dérégulés [Zenz et al., 2005].
La sous-régulation de JunB/AP-1 dans les kératinocytes pourrait donc être un événement initiateur dans l'étiologie du psoriasis, caractérisé par la prolifération de cellules et l'expression dérégulée de cytokines. Des lésions arthritiques avec une pénétrance de 100% ont également été observées, rappelant fortement l'arthrite psoriasique retrouvée chez 5-40% de patients atteints de psoriasis. De plus, la double délétion des deux gènes chez les souris déficiente du gène Rag2 (recombinase activating gene 2) donne un phénotype moins grave avec peu de lésions arthritiques que chez les souris délétées pour Jun et JunB. Ces souris avec knock-out du gène Rag2 ne peuvent plus recombiner les gènes d'immunoglobuline et ne produisent plus de lymphocytes B matures ou de cellules T. Cette étude indique que les lymphocytes T auraient un rôle seulement dans la gravité de la maladie et dans la pathogenèse des lésions arthritiques [Zenz et al., 2005]. Ce phénotype obtenu par le modèle murin de double-délétion de JunB et Jun dans l'épiderme semble donc être le plus ressemblant à celui obtenu chez les humains atteints de psoriasis.
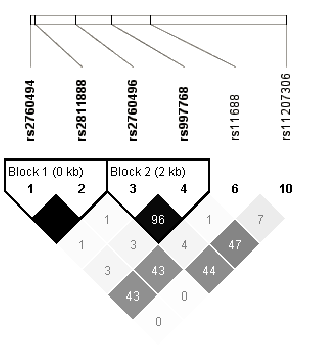
Les protéines Jun et JunB restent des protéines clés et incontournables de la peau. Il est donc utile de choisir Jun et JunB comme gènes candidats dans l'étiologie du psoriasis, d'autant plus que ces gènes, Jun et JunB sont présents dans deux locus de prédisposition au psoriasis, PSORS7 sur le chromosome 1p32 et PSORS6 sur le chromosome 19p13. Pour l'analyse globale de ces gènes, le déséquilibre de liaison entre les SNPs validés dans les bases de données (HapMap/NCBI) et présents dans la région entourant le gène (+10kb) a été étudié. Peu de variants avec une fréquence de l'allèle mineur supérieure à 5% validés dans la population caucasienne ont été identifiés (Figure 3.20Structure DL du gène JUN). L'ensemble des renseignements sur les SNPs sélectionnés ainsi que les résultats obtenus lors de leurs analyses dans le lot I par le programme FBAT individuellement ou conjointement sont décrits dans les tableaux 3.55Identification et test d'association des TagSNPs de JUN, JUNB sur le Lot I par FBAT et 3.56 Analyse d'association des haplotypes de JUN sur le Lot I par FBAT. L'analyse des TagSNPs lors de la stratification des patients du lot I selon la présence ou non de l'haplotype à risque ne donne pas plus de résultats statistiquement significatifs (Table 3.57Analyse d'association des TagSNPs de JUN dans les sous groupes porteurs ou non de l'allèle à risque HLA-Cw6 dans le Lot I).
L'ensemble de ces analyses nous indique que le gène Jun ne semble pas contribuer à la pathogenèse du psoriasis. En revanche, nous ne pouvons pas exclure l'implication de JunB dans le psoriasis. Lors de notre étude sur le gène JunB, seul un variant, présent à l'extérieur du gène, a pu être analysé dû au manque d'informations sur la struture LD du gène. D'après la base de données NCBI, seul le variant rs1061595 est validé dans la population caucasienne et est à une fréquence supérieure à 5%. Il serait donc intéressant de séquencer les régions codantes et régulatrices du gène JunB afin d'identifier les variants qui seront au moins validés dans nos familles françaises. L'analyse de ces variants permettra de nous éclairer sur l'implication de ce gène dans le psoriasis.