


Next: Test d'association basé sur
Up: Méthodes des analyses d'association
Previous: Méthodes des analyses d'association
Contents
La majorité de nos analyses d'association dans les familles ont été réalisées par le test FBAT [Laird et al., 2000]. C'est un test du déséquilibre de transmission (TDT), développé par Spielman et ses collaborateurs (1993), dans lequel les allèles transmis par des parents hétérozygotes aux descendants atteints sont comparées avec la distribution attendue des allèles parmi les descendants. La statitistique générale du test FBAT, nommée 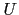 , est donc basée sur une combinaison linéaire des génotypes et des traits des descendants d'une famille:
, est donc basée sur une combinaison linéaire des génotypes et des traits des descendants d'une famille:
![$U=S-E[S], S=\sum_{ij}{T_{ij}X_{ij}}$](img118.png)
où 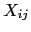 signifie une fonction du génotype du descendant
signifie une fonction du génotype du descendant 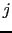 de la famille
de la famille 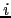 au locus étudié et
au locus étudié et 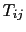 signifie une fonction du trait phénotypique du descendant de la famille .
signifie une fonction du trait phénotypique du descendant de la famille .
Le choix standard est 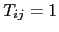 pour les sujets atteints et
pour les sujets atteints et 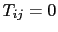 pour les sujets non atteints (dans le cas d'un trait dichotomique). La variance est
pour les sujets non atteints (dans le cas d'un trait dichotomique). La variance est 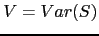 . La forme du test dépend de la codification choisie pour
. La forme du test dépend de la codification choisie pour 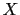 . Si est un scalaire, le test est de la forme:
. Si est un scalaire, le test est de la forme: 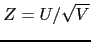 . Sous H0, la statistique
. Sous H0, la statistique 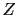 obéit approximativement à une loi normale centrée réduite. Mentionnons qu'en élevant Z au carré, sa distribution peut alors être approximée par une loi du
obéit approximativement à une loi normale centrée réduite. Mentionnons qu'en élevant Z au carré, sa distribution peut alors être approximée par une loi du 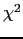 à un degré de liberté. Si est un vecteur, on opte pour un test de la forme suivante:
à un degré de liberté. Si est un vecteur, on opte pour un test de la forme suivante:
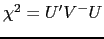 . Sous H0, la statistique obéit approximativement à une loi du à
. Sous H0, la statistique obéit approximativement à une loi du à 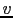 degrés de liberté, où représente le rang de
degrés de liberté, où représente le rang de 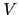 . Ce test basé sur les familles permet, comme décrit dans le chapitre Introduction, d'éviter les biais dus entre autres à la stratification de la population [Lazzeroni and Lange, 1998,Rabinowitz and Laird, 2000].
. Ce test basé sur les familles permet, comme décrit dans le chapitre Introduction, d'éviter les biais dus entre autres à la stratification de la population [Lazzeroni and Lange, 1998,Rabinowitz and Laird, 2000].
De plus, il permet de tester l'association sous deux hypothèses nulles qui sont "non association et non liaison" ou "non association en présence de liaison" (option -e du programme) [Laird et al., 2000]. La première hypothèse a été utilisée pour tester l'association avec les gènes candidats alors que la deuxième a été utilisée pour tester l'association avec les gènes sur les chromosomes 6 et 20, dû à l'existence d'une liaison entre ces régions et la maladie dans nos familles. Dans l'étude des gènes candidats, l'hypothèse nulle "non association et non liaison" est testée car il n'existe aucune preuve de liaison entre les régions étudiées et la maladie dans nos 45 familles françaises [Lesueur et al., 2007a]. Mais contrairement au cas de la deuxième hypothèse, la première entraîne le traitement indépendant des familles nucléaires, qui sont le produit de la décomposition des familles multiplexes.
Le test peut être réalisé selon différents modèles mais le modèle additif, utilisé dans nos études, reste le modèle le plus puissant quel que soit le réel modèle génétique de la maladie [Knapp, 1999,Tu et al., 2000,Horvath et al., 2001]. Il permet aussi d'étudier l'association entre la maladie et des SNPs soit individuellement, soit sous forme d'haplotypes, dans des familles complexes et plus larges que les trios, même dans le cas de génotypes parentaux incomplets [Lange and Laird, 2002]. Il fournit également l'estimation des fréquences alléliques pour chaque marqueur en utilisant les données de génotypage des individus fondateurs des familles (nucléaires ou non). Dans la majorité des cas, les fréquences alléliques de chaque SNP sont proches de celles décrites dans la population CEU du panel HapMap.
Dans le cas des tests d'association sur les gènes candidats, les résultats suggestifs sont déterminés par une valeur de P limite, choisi à P=0.08 pour le premier criblage dans le lot I. En effet, ce seuil est élevé et augmente donc le risque d'obtenir un nombre élevé de faux positifs. Cependant, pour limiter cette dérive, les associations préliminaires sont tentées d'être confirmées par, tout d'abord, une réplication méthodologique, en utilisant une méthode "LNMs" (décrite ultérieurement) puis, une réplication dans un autre lot (Lot 2).
Dans toutes les autres analyses, le seuil limite est de P=0.05. La correction de Bonferroni pourrait être appliquée mais cette correction pour les tests multiples est trop conservative pour ce type d'étude, spécialement quand les SNPs dans les régions candidates sont partiellement corrélés. De plus, les maladies complexes étant les résultantes d'interactions de facteurs environnementaux avec de multiples gènes, qui sont souvent des gènes à effet faible, l'utilisation d'une correction trop conservative pourrait gêner leur identification.
Les analyses sur les haplotypes, constitués de plusieurs SNPs, ont été réalisées en utilisant la fonction HBAT (Haplotype Based Association Test) de FBAT dans les mêmes conditions que pour l'analyse de chaque SNP. Ce test est basé sur l'absence de recombinaison entre les marqueurs qui forment l'haplotype à étudier. Ainsi, la reconstruction des haplotypes est plus facile avec des échantillons familiaux car les haplotypes seront déterminés dans la plupart des cas sans ambiguïté. Cependant, dans le cas où les haplotypes sont difficilement identifiables (cas de données manquantes), ils sont obtenus grâce à l'algorithme EM (Expectation Maximization) [Horvath et al., 2004]. Ceci permet d'identifier une association possible soit avec un marqueur non étudié, mais en déséquilibre de liaison avec un SNP ou une combinaison de SNPs testés, soit avec une combinaison de SNPs. Dans certains cas (ADAM33, CARD15, CYLD, SLC12A8, FLG et STAT3), en raison du nombre élevé de TagSNPs choisis à cause d'un faible niveau de déséquilibre de liaison entre les polymorphismes des gènes en question, le test d'association des nombreux haplotypes qui sont générés à partir des TagSNPs sélectionnés est difficilement réalisable. Nous avons donc décidé de tester la transmission préférentielle aux individus atteints de toutes les combinaisons de 2 ou 3 SNPs possibles. Si une association avec un des haplotypes est observée, un test de permutations est réalisé. La valeur exacte de P du Z est calculée via la méthode de Monte-Carlo pour 1.000.000 de permutations pour chaque haplotype séparément (nommé "1 000 000 permutations P") et pour le test global (nommé "1 000 000 permutations P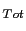 ") dans l'hypothèse d'une non liaison et non association (option -p).
") dans l'hypothèse d'une non liaison et non association (option -p).
Next: Test d'association basé sur
Up: Méthodes des analyses d'association
Previous: Méthodes des analyses d'association
Contents
anouar
2009-08-22