|
Mais D est ťgalement estimť par: |
|
oý et |
 |
population. Ainsi, contrairement au cas oý ![]() , deux SNPs ayant un
, deux SNPs ayant un ![]() de 1 sont totalement ťquivalents et l'un peut Ítre utilisť comme substitut de l'autre (l'information contenue est ťquivalente). Ceci est dŻ ŗ la prťsence de seulement deux des gťnotypes possibles. En pratique, un
de 1 sont totalement ťquivalents et l'un peut Ítre utilisť comme substitut de l'autre (l'information contenue est ťquivalente). Ceci est dŻ ŗ la prťsence de seulement deux des gťnotypes possibles. En pratique, un ![]() de 0,8 est habituellement choisi dans les ťtudes d'association pour dťcrire deux SNPs ťquivalents.
de 0,8 est habituellement choisi dans les ťtudes d'association pour dťcrire deux SNPs ťquivalents.
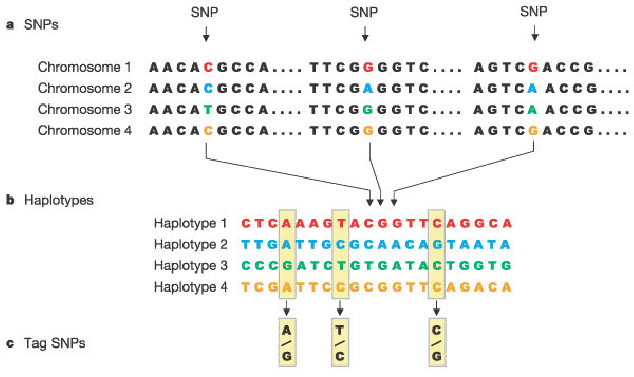
L'ťtude du DL au sein du gťnome montre que le DL peut parfois s'ťtendre sur plusieurs centaines de kilobases. Le gťnome est d'ailleurs structurť en fonction du DL, rťsultant d'une diffťrence du taux de recombinaison le long du gťnome. Cette structuration en blocs de DL peut aussi rťsulter de phťnomŤnes dťpendants de la population ťtudiťe tels que la dťrive gťnťtique (fixation d'un allŤle de maniŤre alťatoire au sein de la population), la croissance et la stratification de la population, la sťlection naturelle et l'apparition de mutations. Le gťnome est donc organisť en blocs de rťgions transmises de maniŤre intacte au cours des gťnťrations. Ces blocs dans lesquels il existe un fort DL et par consťquent, une faible diversitť haplotypique (soit les diffťrentes combinaisons possibles des allŤles de chaque SNP) sont appelťs des blocs de DL (ou blocs haplotypiques). Ainsi, par l'existence de ce fort DL, de nombreux polymorphismes donnent la mÍme information sur la variation gťnťtique au sein du bloc car ils sont souvent hťritťs ensemble. Par consťquent, seulement quelques polymorphismes seront nťcessaires ŗ ťtudier car ils seront reprťsentatifs de l'ensemble des haplotypes existants dans la population, c'est-ŗ-dire de l'ensemble de la variabilitť gťnťtique du bloc. Ils sont nommťs "TagSNPs" (Figure 2.3Approche "TagSNPs" se fait en trois ťtapes).
La caractťrisation des SNPs, en particulier des TagSNPs, a ťtť l'objectif de diffťrents projets d'analyse du gťnome dont HapMap (http://www.hapmap.org/) afin de faciliter les ťtudes d'association [Consortium, 2003]. Ce projet international a permis de cataloguer les variations gťnťtiques (nature, position et frťquence selon les populations) les plus frťquentes chez l'Humain, en analysant l'ADN de 4 populations d'origine africaine, asiatique (japonaise et chinoise) et europťenne [Consortium, 2003]. Cette connaissance permet l'identification de la plupart des haplotypes communs ŗ la population mondiale, variables en frťquence selon les populations. Ainsi, l'ťtude des blocs DL en fonction des populations permet de caractťriser les identificateurs uniques d'un haplotype, les TagSNPs, spťcifique d'une population [Consortium, 2005]. Cela est trŤs utile pour des ťtudes d'association systťmatiques.