


Next: ANALYSE D'ASSOCIATION
Up: Mťthodes des analyses de
Previous: Analyse paramťtrique
Contents
En plus des analyses paramťtriques, diffťrentes analyses non paramťtriques multipoints ont ťtť rťalisťes car ce type d'analyse ne nťcessite pas de connaÓtre les paramŤtres dťjŗ dťcrits prťcťdemment. Une analyse multipoint est plus robuste qu'une analyse bi-point car elle prend en compte les informations obtenues par tous les marqueurs, mÍme ceux qui sont peu informatifs en se basant sur les marqueurs voisins.
Diffťrents programmes permettent de faire ce type d'analyse tels que GENEHUNTER [Kruglyak et al., 1996], Allegro [Gudbjartsson et al., 2000], ou MERLIN [Abecasis et al., 2002]. Cependant, dans le cas de l'ťtude sur des grandes familles, peu de programmes peuvent les analyser ŗ moins d'avoir un nombre limitť de marqueurs tels que LINKAGE [Lathrop et al., 1984] et SIMWALK [Sobel and Lange, 1996]. Pour le premier criblage, deux mťthodes non paramťtriques multipoints ont ťtť utilisťes : la mťthode MLB (Maximum Likelihood Binomial method) [Abel and Muller-Myhsok, 1998] et la mťthode NPL (Non Parametric Linkage). Ces deux mťthodes sont des extensions de la mťthode des paires de germains atteints, dťcrite dans le chapitre Introduction et consistent ŗ dťterminer l'IBD entre tous les germains atteints (dans le cas du MLB) ou entre toutes les paires de germains atteints (dans le cas du NPL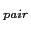 ) ou encore entre tous les malades d'une famille (dans le cas du NPL
) ou encore entre tous les malades d'une famille (dans le cas du NPL ) . Cette approche permet d'accorder plus d'importance ŗ une transmission d'un mÍme allŤle ŗ plusieurs apparentťs atteints qu'ŗ la transmission d'un certain allŤle ŗ une paire de germains atteints.
) . Cette approche permet d'accorder plus d'importance ŗ une transmission d'un mÍme allŤle ŗ plusieurs apparentťs atteints qu'ŗ la transmission d'un certain allŤle ŗ une paire de germains atteints.
Contrairement au NPL qui est basť sur le nombre d'allŤles partagťs IBD par tous les membres atteints d'une famille, la mťthode MLB est basťe sur la distribution binomiale des allŤles parentaux parmi les descendants atteints, considťrant ainsi tous les germains pris comme un entier.
La probabilitť 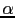 pour un germain atteint de recevoir d'un parent l'allŤle "marqueur" transmis avec l'allŤle "maladie" est ťgale ŗ 0.5 sous l'hypothŤse nulle de non liaison et est supťrieure ŗ 0.5 sous l'hypothŤse de liaison. Le test de liaison est rťalisť ŗ partir d'un test de rapport de vraisemblance
pour un germain atteint de recevoir d'un parent l'allŤle "marqueur" transmis avec l'allŤle "maladie" est ťgale ŗ 0.5 sous l'hypothŤse nulle de non liaison et est supťrieure ŗ 0.5 sous l'hypothŤse de liaison. Le test de liaison est rťalisť ŗ partir d'un test de rapport de vraisemblance
![$\lambda = 2Ln[L(\alpha)/L(\alpha = 0.5)]$](img73.png) avec une statistique distribuťe asymptotiquement comme une distribution mixte de
avec une statistique distribuťe asymptotiquement comme une distribution mixte de
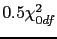 et
et
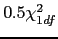 . Le test peut ťgalement Ítre exprimť comme Z
. Le test peut ťgalement Ítre exprimť comme Z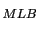 =
=
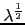 ou LOD MLB =
ou LOD MLB =
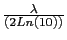 .
.
Ces analyses sont rťalisables gr‚ce aux programmes MLBGH, une extension du programme GENEHUNTER, et MERLIN. Un des inconvťnients de ces programmes est la limite du nombre maximal d'individus pouvant Ítre analysťs. Ainsi, pour permettre l'analyse, les structures familiales ont ťtť modifiťes en les clivant en familles nuclťaires avec le program Mega 2 [Mukhopadhyay et al., 2005]. Les 45 familles ont ťtť divisťes en 77 familles nuclťaires, chacune avec au moins deux germains atteints.
AprŤs l'ťtude de ce premier criblage, un tour du gťnome a ťtť effectuť, non plus sur les 14 premiŤres familles du Lot I mais sur la totalitť des 45 familles du Lot I. Afin de pouvoir comparer avec le premier criblage, nous avons ťtudiť ce deuxiŤme tour de gťnome seul (Criblage 2) ou en ajoutant les marqueurs du premier criblage (Criblage 1+2) en utilisant la mťthode MLB.
Pour l'analyse du deuxiŤme criblage avec ou sans les marqueurs du premier criblage, d'autres mťthodes non paramťtriques multipoints (en particulier en calculant les statistiques NPL et NPL) ont ťtť appliquťes pour l'analyse des familles non fragmentťes gr‚ce au programme SIMWALK2 [Sobel and Lange, 1996]. Contrairement au programme MERLIN, SIMWALK2 utilise une approche MCMC (Markov Chain Monte Carlo) pour estimer les allŤles IBD entre individus atteints selon leur vraisemblance.
La difficultť de ces analyses reste l'estimation du statut IBD des individus. Contrairement aux analyses de paires de germains, l'utilisation de nombreux individus atteints dans l'analyse, permet une meilleure estimation de ce statut, ainsi que l'utilisation simultanťe de l'information de tous les marqueurs sur le chromosome. Nťanmoins, selon la mťthode utilisťe, le moyen de corriger l'effet de la non-indťpendance parmi les germains dans l'analyse est diffťrent. Dans tous les cas, une mauvaise estimation du statut IBD peut diminuer la puissance d'analyse et amener ŗ des faux-positifs. Le seuil de la valeur de P doit Ítre au moins ťgale ŗ 7.4 x 10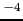 (correspondant ŗ un lod score=2.2) pour indiquer une preuve suggestive d'une liaison, ŗ 2.2 x 10
(correspondant ŗ un lod score=2.2) pour indiquer une preuve suggestive d'une liaison, ŗ 2.2 x 10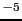 (lod score=3.6) pour une preuve significative et ŗ 3 x 10
(lod score=3.6) pour une preuve significative et ŗ 3 x 10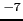 (lod score=5.4) pour une preuve hautement significative [Lander and Kruglyak, 1995]. Cependant, ŗ l'exception de l'ťtude du premier criblage sur 14 familles oý la valeur de P est de 1%, la valeur de P seuil dans l'ensemble des ťtudes de liaison, est de 5% (Z
(lod score=5.4) pour une preuve hautement significative [Lander and Kruglyak, 1995]. Cependant, ŗ l'exception de l'ťtude du premier criblage sur 14 familles oý la valeur de P est de 1%, la valeur de P seuil dans l'ensemble des ťtudes de liaison, est de 5% (Z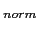 =1.6 pour la mťthode MLB). Conscients des erreurs possibles, nos rťsultats n'ont pas ťtť corrigťs en fonction du nombre de tests effectuťs (Correction de Bonferonni).
=1.6 pour la mťthode MLB). Conscients des erreurs possibles, nos rťsultats n'ont pas ťtť corrigťs en fonction du nombre de tests effectuťs (Correction de Bonferonni).
Next: ANALYSE D'ASSOCIATION
Up: Mťthodes des analyses de
Previous: Analyse paramťtrique
Contents
anouar
2009-08-22